mainmenu
- About Center
- Members
- Research
- Publications
- Facilities
- Super-depth Optical Imaging Lab
- Ultrafast Infrared Spectroscopy Lab
- Ultrafast IR-Visible Spectroscopy Lab
- Instrument and Chemistry Lab
- Molecular Imaging Lab
- Multidimensional Infrared Spectroscopy Lab
- Sample Preparation Lab
- Optical Frequency Comb Spectroscopy Lab
- Single Molecule Imaging Lab
- Spectroscopic Imaging Lab
- Theory and Computation
- Open Access Facility
- Collaborative Research Equipment
- Quantum Spectroscopy Lab
- Equipment Inventory
- Board
As summarized in Sec.3 (Research objectives during the second phase of the CMDS), we plan to carry out researches on the following five different projects:
- · Hydration effects and hydrogen-bonding dynamics
- · Length dependencies of vibrational spectra of polypeptides
- · Vibrational circular dichroism of polypeptides
- · Multidimensional spectrum calculations of lengthy model polypeptides
- · Multidimensional spectrum calculations of natural proteins
5-A. Hydration effects and hydrogen-bonding dynamics
The amide I (mainly C=O stretch of the peptide bond) IR absorption band has been paid much attention since the peak shape and frequency were found to be extremely sensitive to secondary structures of polypeptides and proteins. Furthermore, the hydration of protein was known to play a critical role in stabilizing tertiary structure of a given natural protein and enzyme so that understanding hydration effects and hydrogen-bonding dynamics has to be the first step. In 2001, two-dimensional (2-D) IR pump-probe and photon echo spectroscopic methods were used by Hochstrasser and coworkers and Hamm and coworkers to study vibrational dynamics and spectral evolution of the amide I bands of an N-methylacetamide (NMA), which is a prototype peptide molecule, in an aqueous solution. In order to quantitatively describe these experimental observations, the CMDS has developed several theoretical models and used both quantum chemistry calculation and molecular dynamics simulation methods to predict both 1D and 2D vibrational spectra. We found not only that the predicted spectra were in excellent agreement with experiments but also that the hydrogen-bonding dynamics is ultrafast (both subpicosecond inertial component and picosecond diffusive dynamics) and induces very large solvatochromic frequency shift (~80 cm-1) of the amide I vibration. However, a peculiar experimental observation that the NMA-methanol solution exhibits a doublet IR absorption band has not been fully understood yet. We thus plan to carry out molecular dynamics simulation of NMA-methanol (see the figure on the right) as well as NMA-ethanol solutions to study hydrogen-bonding dynamics of different protic solvents and to elucidate excluded volume effect and competition of the hydrogen-bonding interaction with other van der Waals repulsive interaction in these different protic solvents. Then, we will make direct comparisons with experiments recently carried out by Tokmakoff and coworkers at MIT (U.S.A.). We have made a close collaboration on this particular subject and agreed to carry out this research as a joint project. They have measured 2D IR photon echo spectra of NMA molecule dissolved in a variety of solvents, such as water, methanol, chloroform, etc.. However, due to a lack of theoretical model and computational methods required in the calculation of both 1D and 2D vibrational spectra, they proposed an idea of this strong collaboration between his group at MIT and the CMDS. One of my students, Han who is a Ph.D. student at the CMDS, will make a visit to MIT in the near future and also one of his (Dr. N. Demirdoven) will come visit the CMDS to complete this joint project.
5-B. Length dependencies of vibrational spectra of polypeptides: Gas-phase structure and vibrational properties
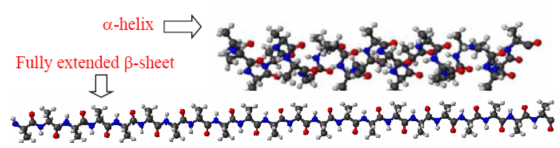
Over the last three years, the CMDS presented theoretical calculation results of polypeptides having two to five amino-acid residues and showed that the vibrational coupling constant does not strongly depend of the chain length and do depend on the secondary structure. Also, the local mode frequencies are dependent on not only its secondary structure but also the position of the peptide bond in a given polypeptide chain. Consequently, we proposed that the site-specific isotope-labeling technique can be used to study local secondary structure around the isotope-labeled peptide bond. A similar idea was tested by Hamm and coworkers in their 2D IR pump-probe study of trialanine in solutions. However, due to their limited ability to quantitatively predict both local mode frequency and inter-peptide coupling constants, their interpretation and its validity remain to be theoretically confirmed. We next plan to carry out extensive quantum chemistry calculation studies (using both ab initio and semiempirical calculation methods) of lengthy a-helical and pleated b-sheet polyalanine molecule having more than 20 residues. As the chain length increases, we expect to see well-behaved trends of the inter-peptide coupling constants and site-independent feature of the local mode frequencies. Furthermore, thus determined parameters will be put into an exciton model to investigate the delocalization of the amide I mode excitons and to find the spectroscopic signatures.
5-C. Vibrational circular dichroism (VCD) of polypeptides
The UV circular dichroism (CD) measurement has been extensively used to estimate the population of a-helix in a given protein. Unlike the UV absorption spectra of different secondary structures, the CD spectrum of a-helix is distinctively different from those of random coil and b-sheet polypeptides. Therefore, using factor analysis of the CD spectrum of an unknown protein, the total content of a-helical amino-acid residues could be measured quantitatively. Recently, the vibrational CD spectroscopy has attracted great attention because it has several advantages in comparison to the electronic CD, i.e., (1) spectral advantages found in IR and Raman spectroscopies, (2) relatively localized transitions (rather than the few, broad, overlapping vibronic bands found in the far UV CD), and (3) measurement of ground electronic state of the molecule (whereas UV CD probes transitions to and vibronic distributions in the excited states). However, due to a lack of proper method that could be used to quantitatively determine local mode frequencies and coupling constants of peptide vibrations, it was impossible to predict VCD band shape and intensity distribution of proteins. Now, using theoretical models developed by the CMDS, we will theoretically study vibrational circular dichroic response of short polypeptides. Also, we plan to purchase a VCD spectrometer and to experimentally measure VCD spectra of various polypeptides.
5-D. Molecular dynamics simulation of acetylproline (model dipeptide) in liquids water and chloroform
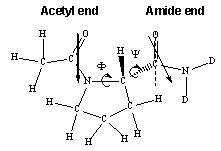
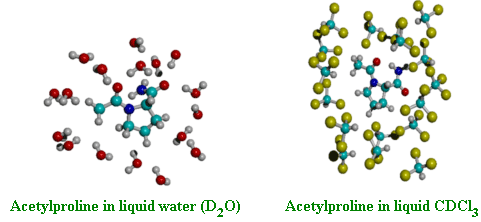
Acetylproline molecule contains two peptide bonds and their relative orientation (thus 3D molecular conformation) is strongly dependent on solvent as well as on the existence of an intramolecular hydrogen bond. The 3D molecular structure is largely determined by the two dihedral angles (F and Y). Hochstrasser and coworkers carried out 2D IR photon echo experiments of this molecule dissolved in liquids water and chloroform. They suggested that there are two distinctively different conformations in chloroform, though there is only one conformer in liquid water. This suggestion has not been confirmed by theoretical investigation yet. We are going to carry out molecular dynamics simulation in combination with extensive ab initio calculations of vibrational frequencies, coupling constants, energies, and hydrogen-bond strengths as functions of the two dihedral angles, F and Y. Then, by comparing our predicted 1D and 2D vibrational spectra with experimental results, it will be possible to address a few unresolved issues (1) how large are the fluctuation amplitudes of the two peptide bonds?, (2) are the two peptide vibrations correlated with each other?, (3) Do the two peptide bonds have different solvation structures?, (4) Is there an intramolecular hydrogen bond when the acetylproline molecule is dissolved in aprotic solvent like chloroform?, and (5) Does the 2D vibrational spectrum provide any novel information on the 3D structure in comparison to the 1D vibrational spectrum?
5-E. Fluctuation, dephasing, and delocalization of vibrational excitons of a-helical polypeptides: 1D and 2D vibrational spectra
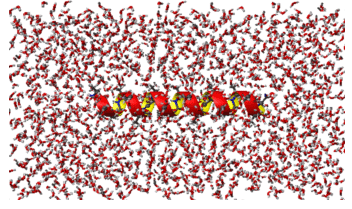
As discussed above in Sec.5-B, we are going to study chain-length-dependencies of various peptide vibrations when the polypeptide forms an extended a-helix conformation. In particular, we will choose polyalanine molecule with 21 alanine residues, since this polypeptide has been known to form a stable a-helix in an aqueous solution. A similar a-helical protein is Fs-helix, which also has 21 residues but three alanine amino-acids are replaced with arginine residues. The 1D and 2D vibrational spectra of Fs-helix have been measured and reported before. However, because its size is very large, it was impossible to do ab initio calculation studies of this system. Furthermore, it is prohibitively difficult and virtually impossible to carry out ab initio molecular dynamics simulation study of this composite system, Fs-helix dissolved in liquid water. Therefore, an alternative method developed by the CMDS over the last three years will be efficiently used to tackle this system to understand fluctuation, dephasing, and delocalization of peptide vibrations of this large system. In the figure below, the ribbon structure of a-helical polyalanine molecule is shown in the middle of periodic box containing thousands of water molecules. Carrying out molecular dynamics simulations, we will be able to measure the conformational fluctuation amplitudes, local hydrogen-bonding dynamics, fluctuation of 20 local mode frequencies, and average and fluctuation amplitude of each vibrational coupling constants of the polyalanine in an aqueous solution
5-F. Multidimensional vibrational spectra of Ubiquitin
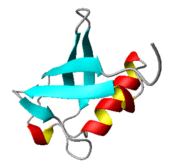
Once we understand the inter-peptide interaction- and solvation-induced vibrational dynamics of model polypeptides (NMA, acetylproline, polyalanine, etc.), we will apply all those theoretical models and methods to study fluctuation dynamics of ubiquitin, which contains 76 amino-acid residues, in an aqueous solution. As can be seen in the ribbon structure, a single ubiquitin protein has both a-helical and b-sheet segments so that it is an ideal but small protein for molecular dynamics simulation studies in combination with quantum chemistry calculation methods. The primary goal of this project is to understand the role of interactions between secondary structures, which stabilize the tertiary structure. We will calculate coherent multidimensional spectra of this protein by taking into account all possible inter-peptide and solute-solvent interactions. Then, we will deliberately remove those interactions making the tertiary structure stable and calculate corresponding spectra. The difference between these two sets of spectra will provide vital information on the spectroscopic signatures of tertiary-structure-dependent spectral changes. Furthermore, by replacing a single segment, either a-helical and b-sheet units, with isotope-exchanged peptide bonds, numerically predicted multidimensional vibrational spectra will reveal the spectroscopic signatures of motifs stabilizing its tertiary structure. Carefully analyzing the spectra, we will be able to theoretically propose novel experiments that can be of use to study protein folding pathway and mechanism.
![]()
Tel : +82-2-3290-4747
Copyright(c) 2014 Center for Molecular Spectroscopy and Dynamics. All Rights Reserved.