mainmenu
- About Center
- Members
- Research
- Publications
- Facilities
- Super-depth Optical Imaging Lab
- Ultrafast Infrared Spectroscopy Lab
- Ultrafast IR-Visible Spectroscopy Lab
- Instrument and Chemistry Lab
- Molecular Imaging Lab
- Multidimensional Infrared Spectroscopy Lab
- Sample Preparation Lab
- Optical Frequency Comb Spectroscopy Lab
- Single Molecule Imaging Lab
- Spectroscopic Imaging Lab
- Theory and Computation
- Open Access Facility
- Collaborative Research Equipment
- Quantum Spectroscopy Lab
- Equipment Inventory
- Board
Before the state-of-the-art theoretical developments are outlined, a series of relevant papers (published before Sep. 2000) are summarized in Table 2.
Table 2. A series of publications by Cho: Theoretical (Before Sep. 2000)
| Title | Journal, vol, page (year) | |
|---|---|---|
| 1 | Off-Resonant Coherent Hyper-Raman Scattering Spectroscopy | J. Chem. Phys., 106, 7550 (1997) |
| 2 | Six-wave mixing spectroscopy: resonant coherent hyper-Raman scattering | J. Chem. Phys. 108, 4013 (1998) |
| 3 | Coherent two-dimensional Raman scattering: frequency-domain measurement of the intra- and intermolecular vibrational interactions | J. Chem. Phys. 108, 1326 (1998) |
| 4 | On the resonant coherent two-dimensional Raman scattering (ReCOTRAS) | J. Chem. Phys. 109, 5327 (1998) |
| 5 | Fifth-order coherent light scattering: Extension of the Kramers-Heisenberg expression for light scattering and two-dimensional measurement of vibrational dynamics | J. Chem. Phys. 109, 6227 (1998) |
| 6 | Time- and frequency-resolved coherent two-dimensional IR spectroscopy: Its complementary relationship with the coherent two-dimensional Raman scattering spectroscopy | J. Chem. Phys. 109, 10559 (1998) |
| 7 | Two-dimensional vibrational spectroscopy | Adv. Multi-photon proc. & spec. 12, 229, (1999) |
| 8 | Two-dimensional vibrational spectroscopy: I. Theoretical calculation of the nonlinear Raman response function of CHCl3 | J. Chem. Phys. 111, 4121 (1999) |
| 9 | Two-dimensional vibrational spectroscopy: II. Ab initio calculation of the coherent 2D IR response function of CHCl3 and comparison with the 2D Raman response function | J. Chem. Phys. 111, 4131 (1999) |
| 10 | Two-dimensional vibrational spectroscopy: III. Theoretical description of the coherent two-dimensional IR-Raman spectroscopy for the investigation of the coupling between both IR- and Raman-active vibrational modes | J. Chem. Phys. 111, 4140 (1999) |
| 11 | Theoretical description of two-dimensional vibrational spectroscopy by infrared-infrared-visible sum frequency generation | Phys. Rev. A61, 23406 (2000) |
| 12 | Intrinsic cascading contributions to the fifth- and seventh-order electronically off-resonant Raman spectroscopies | J. Chem. Phys. 112, 2082 (2000) |
| 13 | Two-dimensional vibrational spectroscopy: IV. Relationship between through-space vibrational coupling and intermolecular distance | J. Chem. Phys. 112, 4553 (2000) |
| 14 | Triply resonant infrared-infrared-visible sum frequency generation: Three-dimensional vibronic spectroscopy for the investigation of the vibrational and vibronic couplings | J. Chem. Phys. 112, 9002 (2000) |
| 15 | Two-dimensional vibrational spectroscopy: V. Novel 2D surface vibrational spectroscopies of adsorbed molecules on surfaces or at interfaces | J. Chem. Phys. 112, 9978 (2000) |
| 16 | Two-dimensional vibrational spectroscopy: VI. Higher-order contributions to the two-dimensional vibrational response functions | J. Chem. Phys. 112, 10496 (2000) |
| 17 | Calculation of the two-dimensional vibrational response function | J. Chem. Phys. 113, 7072(2000) |
| 18 | Two-dimensional vibrational spectroscopy: VII. Investigation of the vibronic and vibrational couplings by using novel triply resonant 2D vibrational spectroscopies | J. Chem. Phys. 113, 7746 (2000) |
2-A. Theoretical: Before the Inauguration of the CMDS (~ Sep. 2000)
The CMDS is the only research group studying coherent multidimensional spectroscopy in Korea. There exist a couple of groups competing with the CMDS in the world. Mukamel at University of Rochester (recently he moved to University of Californian at Irvine) in U.S.A. and Tanimura at the Institute for Molecular Science (he promoted to be a professor at Department of Chemistry of Kyoto University) in Japan have been working on the theoretical investigations of the multidimensional nonlinear spectroscopies. These two investigators theoretically proposed coherent 2D Raman scattering spectroscopy in time-domain in 1993,60,61 and independently Cho and Fleming developed a complete theory on the fifth-order two-dimensional electronic spectroscopy and the main results were published in J. Phys. Chem.(vol. 98, 3478) in 1994. Also, in 1997 Steffen and Duppen at University of Groningen theoretically studied the same 2D Raman scattering process,62,63 which was proposed by Tanimura and Mukamel, later in 1996, but their results are limited to a system with a single vibrational degree of freedom in a weak anharmonicity limit. In parallel with these theoretical works, Saito and Ohmine at Nagoya University carried out the first molecular dynamics simulation of the 2D Raman response functions of liquids water and CS2.64 Fourkas at Boston College and Keyes at Boston University carried out instantaneous normal mode analysis of liquid CS2 to calculate its 1D and 2D Raman responses.65,66 Up until 1997, most works on coherent 2D vibrational spectroscopy focused on the 2D non-resonant (fifth-order) Raman scattering spectroscopy. Since 1997, Cho and coworkers have theoretically proposed numerous novel 2D vibrational spectroscopic methods (see Table 2), which triggered a variety of experimental attempts to test their potential possibilities for future investigations on dynamics and structure determination of complex molecules such as polypeptides in condensed phases, polyatomic molecules, adsorbed molecules on metal surfaces, etc..
2-B. Experimental: Before the Inauguration of the CMDS (~ Sep. 2000)
The first 2D Raman scattering spectroscopy using ultrafast laser pulses were carried out by Tominaga and Yoshihara at Institute for Molecular Science, Japan.67,68 Later, Fleming and Tokmakoff at University of Chicago at that time and Steffen and Duppen showed that the time-resolved fifth-order Raman spectroscopy can be applied to understand the intermolecular vibrational dynamics of liquid CS2 in 1997 and 1998.69,70 However, it was noted that the frequency-domain fifth-order Raman measurement, by Wright and collaborators at the University of Wisconsin at Madison in 1993, was strongly contaminated by two consecutive third-order Raman processes. In relation to this experimental observation, in 1998 Albrecht and coworkers at Cornell University suggested that even the time-domain 2D Raman scattering signal is likely to be the undesired cascading contributions without the direct fifth-order 2D Raman contribution.71,72 Then, Fleming and Blank at University of California at Berkeley showed that the previous 2D Raman signals of CS2 reported by themselves, Yoshihara, and Duppen were largely dominated by the cascading contributions.73 In order to rigorously prove this aspect, Cho, in collaboration with Fleming, attempted to resolve this issue once and for all and provided a systematic theoretical framework on the intrinsic cascading contributions to the electronically non-resonant fifth- and seventh-order Raman scattering processes (J. Chem. Phys. 112, 2082) in 2000. Although Fleming’s group later proved that one can achieve a measurement of the true fifth-order Raman scattering signal (see the figure on the right) by carefully adjusting experimental phase-matching geometry,74,75 this fifth-order Raman scattering method that was theoretically proposed by Tanimura and Mukamel in 1993 has been considered to be extremely difficult and even not to have any bright future as a promising coherent multidimensional vibrational spectroscopy. In contrast, the coherent 2D IR-IR-vis four-wave-mixing (FWM) method proposed by Park and Cho was proven to be free from any cascading contribution and shown to have a strong potential to be the alternative 2D vibrational spectroscopy (see J. Chem. Phys. 109, 10559 (1998)). Wright and coworkers at University of Wisconsin at Madison performed frequency-domain IR-IR-vis FWM experiment of liquid acetonitrile and for the first time they observed a cross peak revealing the existence of vibrational coupling between C-C stretch and CN stretch vibrations.76,77
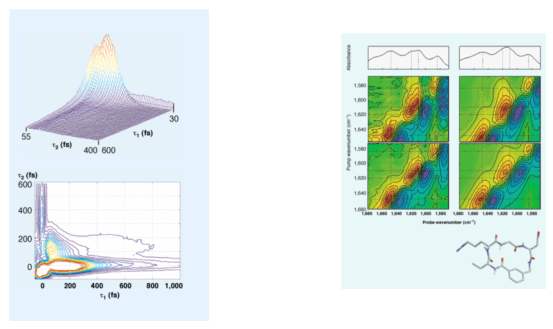
Another class of 2D vibrational spectroscopies based on the IR four-wave-mixing scheme was used by Hochstrasser and coworkers at University of Pennsylvania in 1998.55 In particular, the time- and frequency-resolved IR pump-probe measurements of small polypeptides (2D spectra of cyclic pentapeptide are shown on the right) were performed by his group and the amide I vibrational exciton band was observed. Although, at that time, those experimental results were novel and highly interesting, interpretation of the results was not an easy task because there was no good theoretical model quantitatively describing local amide I mode frequencies and vibrational coupling constants. However, they were able to successfully show that the 2D IR pump-probe and photon echo spectroscopies can be a potentially useful method in studying 3D structures of polypeptides in solutions. It was also noted that these IR-FWM spectroscopic methods are highly analogous to the 2D-COSY NMR spectroscopy.
In Table 3, we summarize a list of theoretical and experimental works, where each of them was considered to be the first attempt in the research field.
Table 3. A summary of the theoretical and experimental works on the coherent multidimensional spectroscopies. (Before Sep. 2000)
| Theoretical | Experimental | Remarks | |
|---|---|---|---|
| 2D Electronic Spectroscopy | · Cho* & Fleming* (1994) | · Joo, Fleming* et.al. (1995) | The first theoretical study of 2D electronic spectroscopy |
| 2D Raman Scattering | · Tanimura & Mukamel* (1993) | · Tominaga and Yoshihara* (1996) · Fleming* & Tokmakoff (1998) · Duppen* & Steffen |
This method turns out not to be useful due to the undesired cascading contributions |
| 2D IR Photon echo & Pump-Probe | · Fayer (1993) · Hochstrasser (1998) |
Reduced 2D IR four-wave-mixing spectroscopy | |
| Coherent 2D IR-IR-vis FWM Spectroscopy | · Cho (1998) | · Wright (1999): frequency-domain · Bonn* and Cho* (2000): |
Four-wave-mixing spectroscopy without any cascading contribution, unlike 2D Raman scattering spectroscopy |
| 2D IR-Raman and Raman-IR | · Cho (1999) | NA | No experiment yet |
| 2D Surface Vibrational Spectroscopy | · Cho (2000) | · Bonn* and Cho* (2000): | First experimental demonstration achieved by Bonn, Cho, and coworkers |
| 3D Vibrational Spectroscopy | · Cho (2000) | NA | Third-, fifth-, and seventh-order NLS. No experimental results yet. |
2-C. Theoretical Investigations (CMDS): After the Inauguration of the CMDS (Oct. 2000 ~ present)
Since Oct. 2000, the CMDS has (1) proposed a number of novel coherent multidimesional spectroscopies and (2) presented quantum chemistry calculation and molecular dynamics simulation results on small molecules like acetonitrile and short polypeptides to search for principle rules governing vibrational interactions and to establish structure-spectrum relationships.
Over the last three years, the CMDS has theoretically proposed numerous novel coherent multidimensional spectroscopic methods and they are summarized in Table 4.
Table 4. Novel coherent multidimensional spectroscopies (CMS) theoretically proposed by the CMDS (after Oct. 2000)
| Spectroscopies | Journal, vol, page (year) | Summary |
|---|---|---|
| IR-OKE | J. Chem. Phys. 114, 9982 (2001) | · Combination of saturation IR pulse and transient Raman measurement · Intermolecular vibrational energy relaxation pathways · Similar to transient nuclear Overhauser enhancement NMR spectroscopy (NOESY) |
| 2-D Fourier deconvolution theory | Chem. Phys. 266, 251 (2001) | · Analytical Fourier deconvolution theory for homodyne detected electronically non-resonant fifth-order Raman scattering signal · Detailed comparisons with experimental results |
| Vibronic FWM | J. Chem. Phys. 114, 8040 (2001) | · Two novel two-dimensional vibrational-electronic spectroscopies |
| 3D spectroscopy | J. Chem. Phys. 115, 4424 (2001) | · The most general theoretical descriptions of three-dimensional spectroscopies · Two-color vibrational photon echo, three-dimensional sum- and difference-frequency generation spectroscopies, and two-color infrared pump–probe spectroscopy |
| 2D vibrational spectroscopy | J. Chem. Phys. 115, 1422 (2001) | · The most general expressions of nonlinear vibrational response functions associated with two-dimensional vibrational spectroscopies |
| Two-color pump-probe | J. Phys. Chem. A, in press (Aug. 2003) | · Two-color transient grating, transient dichroism, and transient birefringence spectroscopies · Two-dimensional line shape and solvation dynamics |
| Time-resolved 2-D VCD | J. Chem. Phys. in press (2003) | · Novel two-dimensional (2-D) circularly polarized (CP) pump-probe (PP) spectroscopy · A combination of IR and VCD (vibrational circular dichroism) measurement in two-dimensional frequency domain |
Hereafter, the principal results of these novel CMS methods will be briefly summarized
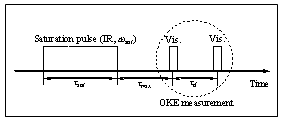
(a) The IR-OKE (IR optical Kerr effect) method was designed to study vibrational energy relaxation from a specific mode, which is excited by a long and intense IR pulse, to solvent bath modes. As demonstrated by numerous workers over the last decade, the optical Kerr effect measurement was found to be an exceptionally useful method for studying intermolecular vibrational dynamics of neat liquids. In particular, the low frequency (less than a few hundreds wavenumber) liquid vibrations can only be studied by the OKE method with a high accuracy. The excess vibrational energy accumulated on a specific mode that is in resonance with the frequency of the saturation IR pulse (see the above figure on the schematic pulse configuration) will dissipate into the solvent modes that are intermolecular in nature. Therefore, by measuring difference between the OKE signal in the presence of the saturation pulse and that in the absence of the saturation pulse as a function of mixing time (tmix), one can elucidate detailed mechanisms and pathways of vibrational energy relaxation process in condensed phases.
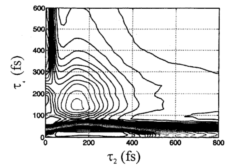
(b) 2-D Fourier deconvolution theory: In collaboration with experimentalists, Fleming and his coworkers at Berkeley, we developed an analytical Fourier deconvolution procedure for homodyne detected electronically non-resonant fifth-order signal that reveals the bare nuclear response function free from the influence of the electronic (hyperpolarizability) responses generated by the five potentially overlapping finite duration pulses used in the experiment. The potential implementation problems with the homodyne deconvolution procedure were evaluated through comparison of the third-order homodyne deconvolution result with that of the well-known third-order heterodyne deconvolution. Then, the homodyne deconvolution is extended to fifth-order where it is used on several measured tensor elements of the direct fifth-order signal. Suggestions were given for improving implementation of the procedure in fifth order so that more information on the direct fifth-order nuclear response as well as the hyperpolarizability responses can be recovered via the deconvolution procedure.
(c) We investigated the effects of temperature on the nonlinear response functions associated with various two-dimensional vibrational spectroscopies. It turns out that the system–bath interaction plays an important role in determining the nature of the temperature-dependencies of the nonlinear response functions and spectra of the two-dimensional vibrational spectroscopy. For a model Hamiltonian, we present exact quantum-mechanical expressions for the nonlinear response functions of two-dimensional vibrational spectroscopies in both the time and frequency domains.
(d) Theoretical descriptions of the three-dimensional spectroscopies were presented by the CMDS by calculating the associated nonlinear response function. The harmonic approximation with the Wick's theorem was previously used to obtain theoretical expressions of the three-dimensional vibrational response functions, where the mechanical and electrical anharmonicities were treated perturbatively. However, the bath-induced memory effect and anharmonicity-induced frequency shift couldn’t be correctly taken into account by the previous theory. By properly taking into account the system–bath interaction, we obtained the most general nonlinear response functions for the three-dimensional vibrational, vibrational-electronic, or electronic spectroscopies. In particular, two-color vibrational photon echo, three-dimensional sum- and difference-frequency generation spectroscopies, and two-color infrared pump–probe spectroscopy were theoretically investigated. This was the last and most complete one of a series of theoretical papers on 3D vibrational & electronic spectroscopies.
(e) Although 2D IR pump-probe spectroscopy has been used to determine precise 3D structure of small peptides in solutions, the relationship between 2D line shape and solvation dynamics was not completely clear. For both two- and three-level systems, we have presented theoretical descriptions of two-color transient grating, transient dichroism, and transient birefringence spectroscopies. The two-dimensional line shapes of these pump-probe spectra were found to be strongly dependent on the solvation dynamics.
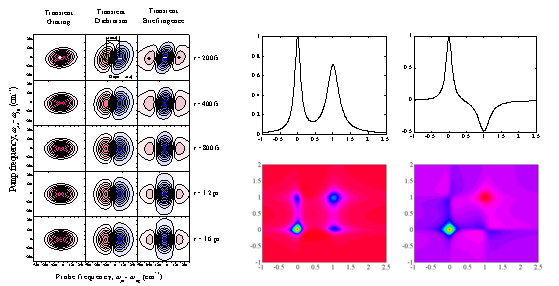
(f) A novel two-dimensional (2-D) circularly polarized (CP) pump-probe (PP) spectroscopy was theoretically studied and proposed by the CMDS. Utilizing circularly polarized pump field, one can measure both the left- and right-CP PP spectra in the 2-D frequency space spanned by the pump and probe field frequencies. It is observed that the relationship between the linearly polarized PP and CP-PP is similar to that between the linear absorption and circular dichroism. Numerically calculated 2-D CP-PP spectra for model systems are presented and compared with the absorption, circular dichroism, and linearly polarized PP spectra (in the above figure, IR absorption (top-left), vibrational circular dichroism (top-right), linearly polarized PP (bottom-left), and circularly polarized PP (bottom-right) spectra are shown).
Next, the state-of-the-art theoretical investigations (by the CMDS) on the vibrational interactions, frequency fluctuation, dephasing, and inhomogeneity of short polypeptides are briefly summarized.
(a) Mono-peptide: The prototype peptide molecule is N-methylacetamide (NMA). For a number of NMA-(D2O)n (n=1~5) clusters, we have carried out extensive
quantum chemistry calculation studies at the Hartree-Fock level to establish a solvation structure-amide I mode frequency relationship.78 Using a multivariate least square regression analysis, we were able to obtain a six-dimensional linear curve that can be used to predict amide I mode (which is
mainly C=O stretch) frequency for an arbitrary configuration of NMA dissolved in liquid water, i.e.,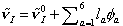 , where
, where 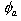 is the electrostatic potential field at the ath site of the NMA molecule and it is produced by the distributed partial charges of the surrounding solvent molecules. We next carried out molecular dynamics simulation of NMA in liquid water, On the basis of molecular dynamics simulation results, both 1D and 2D vibrational spectra of NMA-D2O solution were for the first time calculated and directly compared with experiments.
is the electrostatic potential field at the ath site of the NMA molecule and it is produced by the distributed partial charges of the surrounding solvent molecules. We next carried out molecular dynamics simulation of NMA in liquid water, On the basis of molecular dynamics simulation results, both 1D and 2D vibrational spectra of NMA-D2O solution were for the first time calculated and directly compared with experiments.
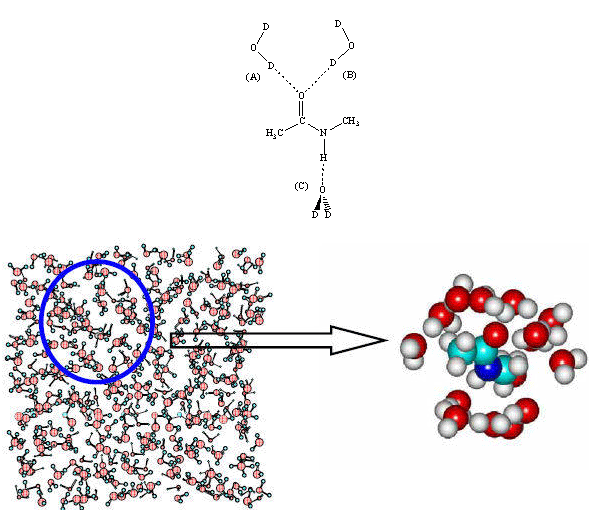
(b) Dipeptides: Motivated by the success of theoretical model for quantitatively predicting solvatochromic vibrational frequency shift and fluctuation amplitude, we next considered glycine dipeptide analog having two peptide bonds.79 Noting that the orientations of the two peptide bonds are determined by two dihedral angles, f and y, and for every 10° of f and y angles ranging from 0 to 360°, we carried out hundreds of ab initio geometry optimizations and vibrational analyses to get the maps (2D contour plots) on local mode frequencies (Fig. (a) and (b)) as well as vibrational coupling constants (Fig. (c) and (d)) in the entire Ramachandran space. These results, which are the far most accurate determination of the vibrational coupling constants depending on the dipeptide 3D conformation, will be used to determine local 3D configuration of a pair of nearest neighboring peptides, once both the local mode frequencies and coupling constants are experimentally determined.
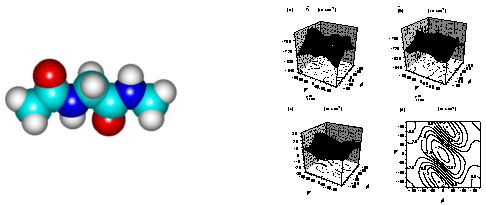
(c) Tripeptides: Tripeptide contains a single peptide surrounded by two peptide groups so that it serves as the smallest and simplest model for studying proteins. As can be seen in the following figure, two pairs of dihedral angles determine the 3D tripeptide conformation. For seven representative conformations, i.e., right-handed a-helix (f = 57, y = -47), 310-helix (-49, -26), left-handed a-helix (60, 60), p-helix (-57, -70), parallel b-sheet (-119, 113), anti-parallel b-sheet (-139, 135), and fully extended b-sheet (180, 180), numerically predicted vibrational spectra were obtained and compared with experiments.80 Furthermore, site-specific isotope (13C or sup>13C18O) labeling effects on amide I infrared absorption spectra were theoretically described and their relationships with 3D conformation were elucidated.
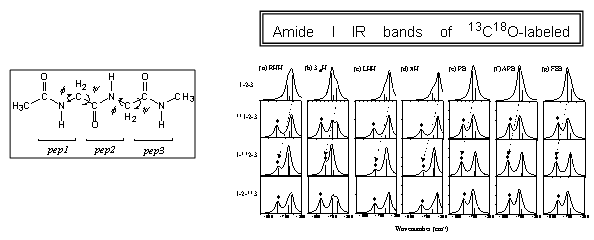
(d) Polypeptides: Varying the number of amino acid residues from 2 to 5, the local amide I mode frequencies and vibrational coupling constants were obtained by using both quantum chemistry calculation method and Hessian matrix reconstruction method, where the latter was recently developed by the CMDS.81 Astonishing results found through this series of investigation are (1) the vibrational coupling constants between two nearest neighboring peptides are strongly dependent on 3D polypeptide conformation, (2) however, they are not sensitively dependent on the chain length, and (3) the local amide I mode frequencies are highly site-dependent, meaning that the local flexibility information can be obtained by the site-specific isotope exchange experiment.
In relation to the current research interests and progresses of the CMDS, Mukamel and coworkers at University of Rochester presented several interesting papers. In J. Chem. Phys. (117, 11089 (2002)), Venkatramani and Mukamel considered the third-order optical response of two coupled anharmonic vibrations interacting with a Brownian oscillator bath that induces energy level fluctuations with arbitrary time scales and degree of correlation. They showed that the two-dimensional correlation plots exhibit distinct signatures of these fluctuations in the various possible three pulse, infrared, femtosecond techniques. In the same journal (118, 3651 (2003)), Moran et al. solved vibrational exciton Hamiltonians for the amide I and amide A modes of both the a- and 310-helical conformations of a fifteen unit polyalanine oligomer constructed using density-functional calculations for smaller model peptides. They calculated energy levels as well as the transition dipoles of all singly and doubly excited-state manifolds and examined a variety of 13C -substituted isotopic derivatives with respect to their ability to reveal differences in local secondary structures in two-dimensional infrared spectra in the amide I region. Dreyer and Mukamel considered full quartic six-mode force field for the NH, CO, and CN stretches of the peptide bonds of a cyclic dipeptide rigidly held by a bridge (2,5-diazabicyclo[2,2,2]octane-3,6-dione) by using ab initio calculation method. The simulation of two-dimensional three-pulse femtosecond infrared measurements were performed and presented the complete set of one- and two-color signals generated at all four possible wavevectors, which shows distinct signatures of anharmonicities, mode couplings, Fermi resonances, and relative transition dipole orientations.
Jansen, Snijders, and Duppen at University of Groningen calculated the third- and fifth-order time-domain Raman responses of liquid carbon disulfide, taking local field effects into account through the dipole-induced dipole approximation to the polarizability (J. Chem. Phys. 114, 10910 (2001). The third-order response was shown to be in excellent agreement with experimental data. The calculated two-dimensional shape of the fifth-order response was compared with recently reported experimental observations of what is claimed to be pure fifth-order response. Considerable discrepancies are observed which might be explained by contamination of the experimental results with sequential and especially parallel third-order cascaded Raman response. A new choice of polarization conditions is proposed, which increases the discrimination against these unwanted cascading effects, as compared to the previously discussed fully polarized and magic angle conditions.
In Phys. Rev. Lett. (85, 1004 (2000)), Ma and Stratt at Brown University carried out theoretical calculations of two-dimensional (fifth-order) Raman scattering spectrum of a liquid. They showed that there exists considerable experimental artifacts and technical difficulties in the state-of-the-art 2D Raman measurement, and reported a new theoretical development: the first microscopic numerical simulation of the 5th-order Raman signal of an atomic liquid Xe. Comparison with an instantaneous-normal-mode treatment, a fully microscopic model which interprets liquid dynamics as arising from coherent harmonic modes, showed that the 5th-order spectrum reveals profound effects stemming from dynamical anharmonicity.
Denny and Reichman at Harvard University presented a fully microscopic molecular hydrodynamic theory for the two-dimensional fifth-order Raman spectrum of an atomic liquid Xe (Phys. Rev. E 63, 65101 (2001). The spectrum was obtained from a simple mode-coupling theory by projecting the dynamics onto bilinear pairs of fluctuating density variables. Good agreement was obtained in comparison with recently reported molecular dynamics simulation results. The microscopic theory provides an understanding of the timescales and molecular motions that govern the two-dimensional signal. Subsequently, predictions were made for the behavior of the spectrum as a function of temperature and density. Their theory showed that novel signatures in the two-dimensional Raman spectrum of supercritical and supercooled liquids are expected. Later, they presented molecular hydrodynamic theory for the fifth-order Raman spectrum obtained from a mode-coupling theory by projecting the dynamics onto bilinear pairs of fluctuating density variables. For the densities and temperatures studied, semi-quantitative agreement was obtained in comparison with molecular dynamics simulation on all time scales. Their theory was contrasted with previous molecular hydrodynamic theories of depolarized light scattering spectra. Extensions of the approach to both classical molecular and quantum liquids were presented (J. Chem. Phys. 116, 1979 (2002)).
2-D. Experimental: After the Inauguration of the CMDS (Oct. 2000 ~ present)
There have been numerous reports on applications of coherent multidimensional spectroscopies to study structure and dynamics of short polypeptides over the last three years.
Woutersen and Hamm at Max Born Institute carried out polarization sensitive two-dimensional (2D) vibrational spectroscopic experiment on the amide I mode of trialanine in aqueous solution to determine its backbone structure (J. Phys. Chem. B 104, 11316 (2000)). Exploiting polarization sensitivity of the 2D pump-probe signal, they showed that the cross-peak structure hidden under the strong diagonal peaks can be selectively measured. The two dihedral angles f and y characterizing the peptide backbone structure were derived directly from the cross-peak intensity and anisotropy, demonstrating the potential of 2D spectroscopy as a tool for peptide structure elucidation. In J. Chem. Phys. (115, 7737 (2001)), they applied nonlinear two-dimensional vibrational spectroscopy to investigate the amide I band of an alanine-based 21-residue a-helical peptide in an aqueous solution. Although the linear absorption spectrum consists of a single, broad amide I band, the 2D vibrational spectrum clearly revealed that this band is composed of two amide I transitions, which are assigned to the A and E1 modes. The A–E1 frequency splitting is found to be approximately 10 wavenumber. They further found that the amide I band is inhomogeneously broadened due to conformational disorder of the helix. The 2D line shapes can be well described using distributions of the dihedral angles f and y around their average values with a width of 20°, confirming previous molecular-dynamics studies. Their time-resolved 2D measurements show that the conformation fluctuates on a time scale of picoseconds. In 2002 (J. Chem. Phys. 117, 6833), Hamm, Stock, and coworkers used nonlinear time-resolved vibrational spectroscopy to compare spectral broadening of the amide I band of the small peptide trialanine with that of N-methylacetamide, a commonly used model system for the peptide bond. In contrast to N-methylacetamide, the amide I band of trialanine is significantly inhomogeneously broadened. Employing classical molecular-dynamics simulations combined with density-functional-theory calculations, they investigated the origin of the spectral inhomogeneity. While both systems exhibit similar hydrogen-bonding dynamics, it was found that the conformational dynamics of trialanine causes a significant additional spectral broadening. In particular, transitions between the polyGly-II and the right-handed a-helix conformations are identified as the main source of the additional spectral inhomogeneity of trialanine. The experimental and computational results suggest that trialanine adopts essentially two conformations: PII (~80%) and aR (~20%). Subsequently, they studied the amide I mode of trialanine and two of its isotopomers dissolved in heavy water (J. Chem. Phys. 114, 2727 (2001). In particular, they used site-directed 13C isotope substitution to change the individual frequencies of the coupled oscillators, and hence to modify specific matrix elements of the molecular Hamiltonian. They found that all of the results could be well described by an excitonic model for the amide I band, using the same coupling strength and dipole–dipole angle for all three isotopomers.
Zanni, Asplund, and Hochstrasser at University of Pennsylvania carried out the stimulated infrared photon echo measurement of N-methylacetamide-D (NMAD). The correlation function was modeled as a single exponential plus a
constant, and it was found that most of the NMAD vibrational frequency distribution is motionally narrowed with a pure dephasing time of 1.12 ps. The two-dimensional infrared 2D IR spectrum of NMAD was also obtained by heterodyning the echo field with a weak local oscillator pulse. The real and imaginary portions of the 2D IR spectrum exhibit multiple peaks due to 0–1 and 1–2 coherences that are excited, which were not resolved in the absolute magnitude of the 2D IR spectrum. Using the correlation function determined from the stimulated photon echo, the 2D IR spectrum was accurately simulated.
Resolution enhancement of the 2D IR spectrum was performed by manipulating the photon echo field with window functions. The enhanced experimental and simulated 2D IR spectra are dramatically narrowed. Later, they carried out heterodyned femtosecond infrared two-pulse and three-pulse photon echo measurements of the dipeptide acetylproline-NH2 in D2O and CDCl3 and the results were compared with force field calculations of the peptide structures (J. Phys. Chem. B 105, 6520 (2001)). The heterodyned two-dimensional infrared (2D IR) spectra obtained from the measurements exhibit diagonal peaks and cross-peaks that are determined by the structures and vibrational dynamics of the cetylproline-NH2 molecule. In CDCl3, the 2D IR spectra from the two-pulse experiments resolved two acetyl amide I bands and two amino amide I bands that are not resolved in the linear spectrum. Thus, acetylproline-NH2 must have at least two structures in CDCl3. The angles between the amide I transition dipoles of the structures were determined to be <20° and 35° from polarized 2D IR measurements. A single structure is found in D2O with an angle of <20°. In the three-pulse experiments, the cross-peak intensities and polarizations were found to change with the waiting time between the second and third pulses. By comparison to the changes in the diagonal peaks, these effects are attributed to population and coherence transfer processes. The vibrational dynamics and inhomogeneous distributions at the acetyl and amino ends of acetylproline-NH2 are observed to be different. In 2003 (J. Chem. Phys. 118, 7733), Rubtsov, Wang, and Hochstrasser showed that dual frequency two-dimensional infrared spectroscopic measurement is feasible using three IR fields, two at 3 and one at 6 mm from two pulses, creating a joint nonlinear response from both the amide-A (N-H stretch) and the amide-I transitions of two model peptides AcAlaOMe and caprolactam. The 2D-IR spectra yield anharmonicities and correlations of inhomogeneous distributions. A positive correlation between the fundamental N–H mode frequency and its anharmonicity is found for the dipeptides. The amide-I frequencies are correlated with N–H mode frequencies and anti-correlated with the amide-I/N–H mode coupling. Structural implications of these results are discussed in relation to the anharmonic potential surface.
Tokmakoff and coworkers at MIT showed that the two-dimensional infrared photon echo spectroscopy can be used to describe the anharmonic nuclear potential of two coupled molecular vibrations (Phys. Rev. Lett. 86, 2154 (2001)). The two-dimensional spectrum showed diagonal and off-diagonal features, each composed of two peaks. The splitting between these peaks is directly related to the anharmonicity, while the relative amplitude of the diagonal and off-diagonal features describes the projection angle between interacting dipoles. In J. Chem. Phys. (115, 10814 (2001)), they used two-dimensional vibrational spectroscopy to characterize transient molecular structure by measuring the couplings and projection angles between two strongly coupled anharmonic vibrations. Two-dimensional Fourier-transform infrared spectra of the coupled carbonyl stretches of Rh(CO)2(C5H7O2) in hexane were obtained from femtosecond vibrational echo signals detected with spectral interferometry. The eight resonances in the two-dimensional spectrum can be interpreted as two diagonal peaks and two cross peaks, each split into a pair. The splitting between the peak pairs is directly related to the diagonal and off-diagonal anharmonicity of the symmetric and asymmetric carbonyl stretches. The ratio of the amplitude of the cross peaks for two different polarization geometries determines the projection angle between the coupled transition dipoles. The experimental characterization of the vibrational eigenstates allows the local carbonyl structure to be modeled as bilinearly coupled cubic anharmonic oscillators. The interaction between the carbonyl stretches arises from the mutual bonding with the rhodium metal center. This two-dimensional infrared experiment characterizes the structure with a time window of roughly 20 ps, suggesting a general method for capturing transient molecular structure in solution. In Phys. Rev. Lett. (90, 47401 (2003)), Tokmakoff and coworkers measured absorptive line shapes in two-dimensional infrared (2D IR) vibrational spectra that are important for an intuitive interpretation of molecular structure and dynamics. The 2D correlation spectrum of a coupled vibrational system reveals certain spectral features with tilted line shapes that are explained in terms of unequal contributions of Louville-space pathways.
Zhao and Wright at University of Wisconsin, Madison, used IR-IR-vis four-wave-mixing scheme which is an analog to 2D NMR and demonstrated its spectral selectivity, sensitivity to the interactions causing mode coupling, and ability to spectrally resolve isotopic mixtures of acetonitrile (CH3CN) solutions (Phys. Rev. Lett. 84, 1411 (2000)). The method was shown to discriminate against uncoupled vibrational modes and isolates the features that are associated with intra- or intermolecular interactions. However, they were not able to interpret the peculiar asymmetric 2D line shape of the cross peak revealing coupling between the CN stretch and C-C stretch vibrations. Later, in 2002, Kwak, Cha, and Cho at the CMDS investigated the origin of the vibrational coupling that was observed between the CC and CN stretching modes of acetonitrile by doubly vibrationally enhanced IR–IR–vis four-wave-mixing spectroscopy by carrying out various ab initio calculations including DFT (B3LYP), HF, and MP2 methods with the same baisis set, 6-311++G** (J. Chem. Phys. 117, 5675 (2002)). We calculated the linear and nonlinear susceptibilities of the combination bands and cross peaks and compared with the experimental values reported by Zhao and Wright. The agreement between ab initio results and experiments was quantitative. By separately analyzing the contributions from each coherence pathway to the vibrational coupling of the CC and CN stretching modes, a quantitative understanding of this 2D IR-IR-vis four-wave-mixing signals was possible. Although the direct coupling of the CC and CN stretching modes by mechanical and electric anharmonicity coupling is sizable, the CH bending and CH stretching modes are also involved in the vibrational coupling between CC and CN stretching modes as promoting modes. The numerically simulated two-dimensional spectrum for CH3CN liquid was presented and compared with experiment. It was found that the interference among different pathways plays a central role in describing the distorted, asymmetric shape of the 2D IR-IR-vis four-wave-mixing spectrum. In addition, the cross peak associated with the vibrational coupling between the CH and CN stretching mode is also calculated and its magnitude is compared with that of the CC and CN stretching modes. It is believed that this paper with Wright being a co-author was the first one demonstrating how the quantum chemistry calculation methods can be of use to quantitatively predict and describe 2D vibrational spectroscopic observable of small molecules like acetonitrile and addressed an important issue of vibrational coupling processes involved in coherent multidimensional spectroscopy.
![]()
Tel : +82-2-3290-4747
Copyright(c) 2014 Center for Molecular Spectroscopy and Dynamics. All Rights Reserved.