mainmenu
- About Center
- Members
- Research
- Publications
- Facilities
- Super-depth Optical Imaging Lab
- Ultrafast Infrared Spectroscopy Lab
- Ultrafast IR-Visible Spectroscopy Lab
- Instrument and Chemistry Lab
- Molecular Imaging Lab
- Multidimensional Infrared Spectroscopy Lab
- Sample Preparation Lab
- Optical Frequency Comb Spectroscopy Lab
- Single Molecule Imaging Lab
- Spectroscopic Imaging Lab
- Theory and Computation
- Open Access Facility
- Collaborative Research Equipment
- Quantum Spectroscopy Lab
- Ultrafast Raman Probe Spectroscopy Lab
- Equipment Inventory
- Board
Posters and Short Descriptions
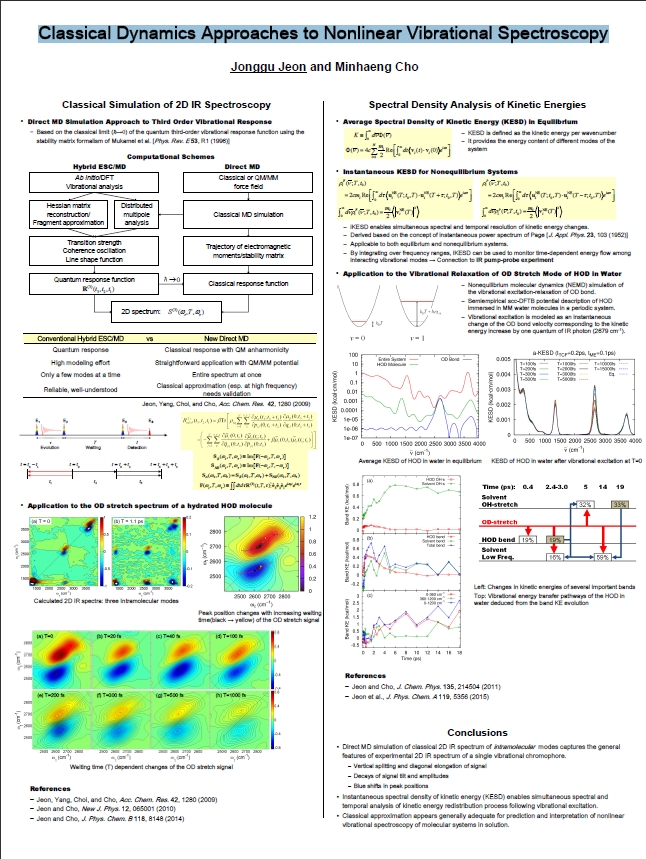
Classical Dynamics Approaches to Nonlinear Vibrational Spectroscopy
Summary:
My research centers on the theoretical study of molecular systems under the classical approximation in relation with nonlinear vibrational spectroscopy. Here, I'd like to introduce two examples under this theme.
To theoretically describe 2D IR spectroscopy, which elucidates molecular structure and dynamics, we need to calculate the third order vibrational response function. The present research aims to calculate this response function in the classical limit by way of molecular dynamics (MD) simulation employing quantum mechanical/molecular mechanical (QM/MM) force field. In this approach, important characteristics of molecular systems, such as potential anharmonicity, spectral diffusion, chemical exchange, and vibrational energy relaxation are naturally reflected in the calculated spectrum without further theoretical modeling. Moreover, the spectra spans the entire molecular vibrational frequency range, revealing correlations between disparate vibrational modes.
The time-dependent changes of kinetic energy can be analyzed in the frequency domain using the spectral density of kinetic energies (KESD) which provides essential information for the vibrational energy relaxation processes. I have developed KESD appropriate for nonequilibrium dynamics study based on velocity time correlation function. The method has been applied to vibrational energy relaxation of the OD stretch mode in water and it was possible to map out the intra- and intermolecular energy transfer pathways of the system. The method is quite general and can be used with more sophisticated force field models.
![]()
Tel : +82-2-3290-4747
Copyright(c) 2014 Center for Molecular Spectroscopy and Dynamics. All Rights Reserved.